Pipeline Summary¶
The Stenglein Lab Standard Taxonomic Assessment Pipeline is a metagenomics pipeline that taxonomically assigns reads in a NGS dataset that remain after filtering host sequences.
Input¶
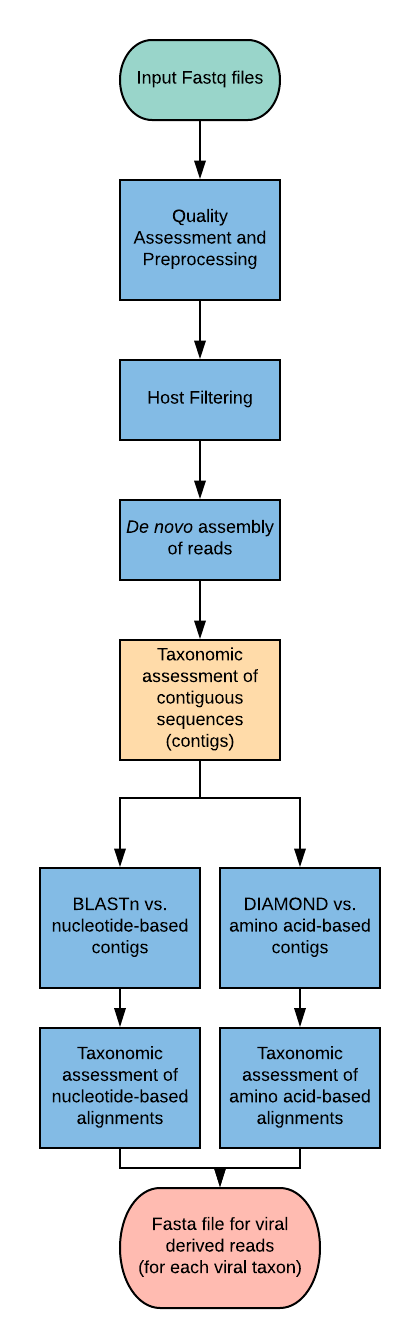
This version of the Stenglein Lab Standard Taxonomic assessment pipeline requires the input of paired FASTQ files. Specific input instructions are explained in detail in the Running the Pipeline section.
Quality Assessment and Pre-Processing¶
The input files are first assessed using FastQC (version 0.11.5) to perform an initial quality control inspection on the raw data [1]. Sequences are then trimmed using Cutadapt (version 1.9.1) to trim low-quality bases and adaptor sequences [2]. Default pipeline parameters set quality base to 33 and quality cutoff to 30 for the 50 and 30 ends. The first base of each sequence is also trimmed. The sequence clustering tool CD-HIT (version 4.6.5) is then used to collapse reads with 99% global pairwise identity, leaving only unique reads for further analysis [3]. Following these pre-processing steps, a second quality control analysis is run using FastQC.
Host Filtering¶
Host-derived sequences are then filtered using the Bowtie2 alignment tool (version 2.2.5). First, a bowtie index is generated from the host genomic sequence and then sequences aligning with a local mode alignment score greater than 60 are removed [4].
De novo Assembly of Reads¶
SPAdes genome assembler (version 3.5.0) is used to generate contiguous sequences (contigs) of non-host reads [5].
Taxonomic Assessment of Contigs¶
To taxonomically categorize sequences, the NCBI nt database is queried with all contigs using the BLASTn alignment tool (version 2.2.30+) [6,7]. Any hit with an expect value less than 10^8 is assigned taxonomically according to the sequence with the highest alignment score [6,8]. To attempt to categorize contigs that are too divergent to produce a high scoring nt–nt alignment, the NCBI nr database is queried in a DIAMOND (version 2.23) search with a minimum length of 20 amino acids and an expect value of 0.01 [9].
Output¶
The output of this metagenomics pipeline is … which is described in more detail in the Example Analysis and Output section.